Step 2: performing the region- or gene-based association tests
- The commands are the same as the step 2 for single-variant assoc tests, except that
- A group file is specified (–groupFile), which contains the genetic marker IDs, annotations and weights (if any) for each set to be tested
- Allow for multiple maskes for each set (gene or region)
- Use –annotation_in_groupTest to list different annotations, seperated by comma. Within each annotation combination, annotations are seperated by ‘:’
- e.g. “lof,missense:lof,missense:lof:synonymous” is to test lof only, missense+lof, and missense+lof+synonymous
- Use –maxMAF_in_groupTest for different max MAF cutoffs seperated by comma
- e.g. 0.0001,0.001,0.01 (default)
- Use –maxMAC_in_groupTest for different max MAC cutoffs seperated by comma
- e.g. 1,2 for singletons and doubletons
- by default, this option is not applied
- the max MAC will be converted to max MAF and merged to –maxMAF_in_groupTest in the analysis
- In the example, there will be 9 masks applied to each set and the 9 p-values will be combined based on the Cauchy combination
- Use –annotation_in_groupTest to list different annotations, seperated by comma. Within each annotation combination, annotations are seperated by ‘:’
- By default, SKAT-O test will performed (with BURDEN and SKAT test results output too). Use –r.corr=1 to only perform BURDEN test
- If SKAT-O tests are performed (–r.corr=0), the single-variant assoc tests results are also output
- If only BURDEN tests are performed (–r.corr=1), the single-variant assoc tests are not performed by default. Use –is_single_in_groupTest=TRUE to output the single-variant assoc tests results.
- Use –is_output_markerList_in_groupTest=TRUE to output marker list included in each test.
- By default, the program first check if per-marker-weight is provided in the group file. If not, the program calcuates weights based on MAF from the Beta distribution with paraemters weights.beta. Use –is_no_weight_in_groupTest=TRUE to not use any weights in the tests.
- The cutoffs for single markers specified using –minMAF, –minMAC, and –minInfo are also applied in the region/set-based tests
- Same as the single-variant association tests, conditional analysis based summary stats can be performed (–condition)
WARNING
- For step 1 with version < 1.0.6
- If the variance ratios were estimated using a full GRM and a sparse GRM,specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2
- If the variance ratios were estimated using a sparse GRM and null GRM, do not specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2
- If no variance ratios were estimated and a spare GRM was used for fitting the null model, do not specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2
- For step 1 with version >= 1.0.6
- if sparse GRM was used in step 1, do specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2
- If a a sparse GRM was used for fitting the null model and variance ratios were estimated with sparse and null GRMs, in Step 2, the sparse GRM (–sparseGRMFile, –sparseGRMSampleIDFile) and variance ratios (–varianceRatioFile) are used as input. Use –is_output_markerList_in_groupTest=TRUE to output the markers used for each test.
- –LOCO=FALSE
- WARNING ** If step 1 was generated with version < 1.0.6, DO NOT specify –sparseGRMFile and –sparseGRMFile ** If step 1 was generated with version >= 1.0.6. Use –is_fastTest=TRUE for the fast test and specify –sparseGRMFile and –sparseGRMFile
Rscript step2_SPAtests.R \ --bgenFile=./input/genotype_100markers.bgen \ --bgenFileIndex=./input/genotype_100markers.bgen.bgi \ --SAIGEOutputFile=./output/genotype_100markers_bgen_groupTest_out.txt \ --chrom=1 \ --AlleleOrder=ref-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_sparseGRM.rda \ --varianceRatioFile=./output/example_binary_sparseGRM.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest="lof,missense:lof,missense:lof:synonymous" \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --is_output_markerList_in_groupTest=TRUE \ --LOCO=FALSE \ --is_fastTest=TRUE -
If a sparse GRM was used for fitting the null model and no variance ratios were estimated, in Step 2, use the same sparse GRM (–sparseGRMFile, –sparseGRMSampleIDFile) as input
Rscript step2_SPAtests.R \ --bgenFile=./input/genotype_100markers.bgen \ --bgenFileIndex=./input/genotype_100markers.bgen.bgi \ --SAIGEOutputFile=./output/genotype_100markers_bgen_groupTest_out_sparseGRMforStep1.txt \ --chrom=1 \ --AlleleOrder=ref-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_sparseGRM.rda \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --LOCO=FALSE - If a full GRM was used in Step 1 for fitting the null model and variance ratios were estimated with full and sparse GRMs, in Step 2, the sparse GRM (–sparseGRMFile, –sparseGRMSampleIDFile) and variance ratios (–varianceRatioFile) are used as input. Use –is_output_markerList_in_groupTest=TRUE to output the markers used for each test.
- –LOCO=TRUE
- If step 1 was generated with version >= 1.0.6. Use –is_fastTest=TRUE for the fast test
Rscript step2_SPAtests.R \ --bgenFile=./input/genotype_100markers.bgen \ --bgenFileIndex=./input/genotype_100markers.bgen.bgi \ --SAIGEOutputFile=./output/genotype_100markers_bgen_groupTest_out.txt \ --chrom=1 \ --LOCO=TRUE \ --AlleleOrder=ref-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_fullGRM.rda \ --varianceRatioFile=./output/example_binary_fullGRM.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest="lof,missense:lof,missense:lof:synonymous" \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --is_output_markerList_in_groupTest=TRUE \ --is_fastTest=TRUE -
Only perform BURDEN test with –r.corr=1. Use –minGroupMAC_in_BurdenTest for the minimum MAC of the testing “burden marker” in the Burden test. NOTE: the sparse GRM is not required. If only perform BURDEN tests, the Step 1 output generated for the single-variant assoc tests can be re-used for BURDEN tests as the variance ratios are estimated using full GRM and null GRM
Rscript step2_SPAtests.R \ --bgenFile=./input/genotype_100markers.bgen \ --bgenFileIndex=./input/genotype_100markers.bgen.bgi \ --SAIGEOutputFile=./output/genotype_100markers_bgen_groupTest_out_onlyBURDEN.txt \ --chrom=1 \ --LOCO=TRUE \ --AlleleOrder=ref-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary.rda \ --varianceRatioFile=./output/example_binary.varianceRatio.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest="lof,missense;lof,missense;lof;synonymous" \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --r.corr=1 -
Use –condition= to perform conditioning analysis
Rscript step2_SPAtests.R \ --bgenFile=./input/genotype_100markers.bgen \ --bgenFileIndex=./input/genotype_100markers.bgen.bgi \ --SAIGEOutputFile=./output/genotype_100markers_bgen_groupTest_out_cond.txt \ --chrom=1 \ --LOCO=TRUE \ --AlleleOrder=ref-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_fullGRM.rda \ --varianceRatioFile=./output/example_binary_fullGRM.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --condition=1:30:A:C,1:79:A:C - Per-marker weights are included in the group file –groupFile=./input/group_new_chrposa1a2_withWeights.txt
- WARNING ** If step 1 was generated with version < 1.0.6 ** if sparse GRM was used for variance ratio estimation, do specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2, otherwise, do NOT specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2 ** If step 1 was generated with version >= 1.0.6 ** if sparse GRM was used in step 1, do specify –sparseGRMFile and –sparseGRMSampleIDFile in step 2
Rscript step2_SPAtests.R \ --bgenFile=./input/genotype_100markers.bgen \ --bgenFileIndex=./input/genotype_100markers.bgen.bgi \ --SAIGEOutputFile=./output/genotype_100markers_bgen_groupTest_out_sparseGRMforStep1_withWeights.txt \ --chrom=1 \ --AlleleOrder=ref-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_sparseGRM.rda \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2_withWeights.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --LOCO=FALSE \ --varianceRatioFile=./output/example_binary_sparseGRM.varianceRatio.txt \ --is_fastTest=TRUE - Use PLINK file as input for genotypes/dosages (–bedFile=, –bimFile=, –famFile=, –AlleleOrder=)
- –AlleleOrder can be alt-first or ref-first. It has to be correctly specified, otherwise, the variants in the PLINK file will not be matched with the markers in the groupFile
Rscript step2_SPAtests.R \ --bedFile=./input/genotype_100markers.bed \ --bimFile=./input/genotype_100markers.bim \ --famFile=./input/genotype_100markers.fam \ --SAIGEOutputFile=./output/genotype_100markers_plink_groupTest_out.txt \ --chrom=1 \ --LOCO=TRUE \ --AlleleOrder=alt-first \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_fullGRM.rda \ --varianceRatioFile=./output/example_binary_cate.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 - VCF file as input
- –vcfFileIndex takes .csi index file as input, which can be generated using tabix –csi -p vcf ./input/genotype_100markers.vcf.gz
- –vcfField is GT or DS
- –chrom doe not need to be specified for VCF files for set-based tests. But it must be specified if LOCO=TRUE. –chrom must be the same as the CHROM string in the VCF file
- chr in marker IDs needs to be matched the CHROM string, e.g. ./input/group_new_chrposa1a2_withchr.txt
#CHROM in VCF is 1 Rscript step2_SPAtests.R \ --vcfFile=./input/genotype_100markers.vcf.gz \ --vcfFileIndex=./input/genotype_100markers.vcf.gz.csi \ --vcfField=GT \ --SAIGEOutputFile=./output/genotype_100markers_vcf_groupTest_out.txt \ --LOCO=FALSE \ --chrom=1 \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_fullGRM.rda \ --varianceRatioFile=./output/example_binary_cate.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 ##CHROM in VCF is chr1, --LOCO=FALSE so no --chrom is needed. Rscript step2_SPAtests.R \ --vcfFile=./input/genotype_100markers_missGT.withchr.vcf.gz \ --vcfFileIndex=./input/genotype_100markers_missGT.withchr.vcf.gz.csi \ --vcfField=GT \ --SAIGEOutputFile=./output/genotype_100markers_vcf_groupTest_out.txt \ --LOCO=FALSE \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_fullGRM.rda \ --varianceRatioFile=./output/example_binary_cate.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2_withchr.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 ##CHROM in VCF is chr1, --LOCO=TRUE so --chrom=chr1 Rscript step2_SPAtests.R \ --vcfFile=./input/genotype_100markers_missGT.withchr.vcf.gz \ --vcfFileIndex=./input/genotype_100markers_missGT.withchr.vcf.gz.csi \ --vcfField=GT \ --SAIGEOutputFile=./output/genotype_100markers_vcf_groupTest_out.txt \ --LOCO=TRUE \ --minMAF=0 \ --minMAC=0.5 \ --sampleFile=./input/samplelist.txt \ --GMMATmodelFile=./output/example_binary_fullGRM.rda \ --varianceRatioFile=./output/example_binary_cate.varianceRatio.txt \ --sparseGRMFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx \ --sparseGRMSampleIDFile=output/sparseGRM_relatednessCutoff_0.125_1000_randomMarkersUsed.sparseGRM.mtx.sampleIDs.txt \ --groupFile=./input/group_new_chrposa1a2_withchr.txt \ --annotation_in_groupTest=lof,missense:lof,missense:lof:synonymous \ --maxMAF_in_groupTest=0.0001,0.001,0.01 \ --chrom=chr1
Input files
- Please Refer to the SAIGE (Single-variant test) Step 2 input files for details. In addition, there are additonal input files required for the set-based tests
-
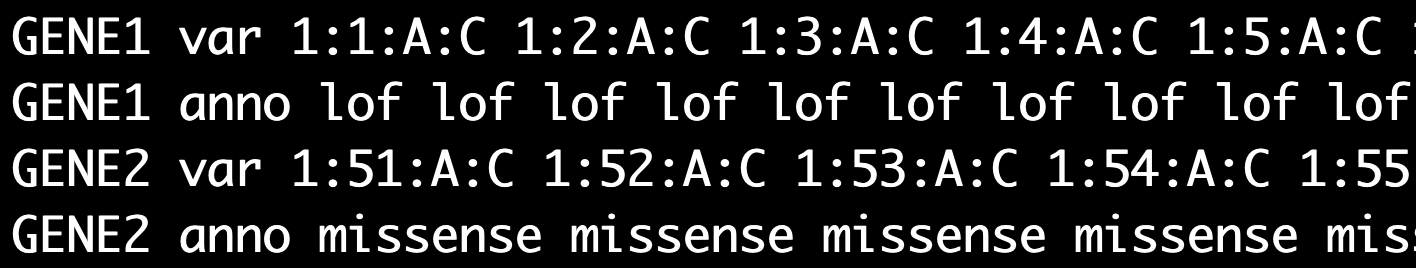
(Required. Specific for set-based tests)** Group file containing marker IDs, annotations, and/or weights for each set (gene or region). * The first column contains the set name. * Group file has 2 or 3 lines for each set. * marker IDs and annotations are required and weights are optional. * The second column has var (indicating the line is for marker IDs), anno (indicating the line is for annotations), and weight (indicating the line is for weights of markers used in the set-based tests)
* If weights are not included in the group file, the weights will be calcuated as beta(MAF, 1, 25) by default- group file without weights
less -S ./input/group_new_chrposa1a2.txt
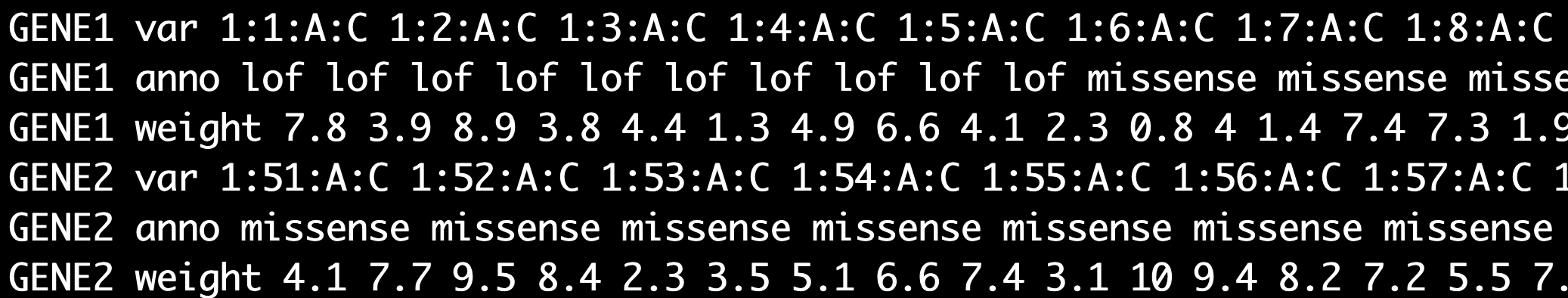
* group file with weights
```
less -S ./input/group_new_chrposa1a2_withWeights.txt
```
Output files
-
A file with region- or gene-based association test results
head -n 1 ./output/genotype_100markers_bgen_groupTest_out_cond.txt

Region: set name
Group: annotation mask
max_MAF: maximum MAF cutoff
Pvalue: p value for SKAT-O test
Pvalue_Burden: p value for BURDEN test
Pvalue_SKAT: p value for SKAT test
BETA_Burden: effect size of BURDEN test
SE_Burden: standard error of BETA_Burden
#if --condition= is used for conditioning analysis, the conditional analysis results will be output
Pvalue_cond, Pvalue_Burden_cond, Pvalue_SKAT_cond, BETA_Burden_cond, SE_Burden_cond
MAC: minor allele count in the set
MAC_case: minor allele count in cases
MAC_control: minor allele count in controls
Number_rare: number of markers that are not ultra-rare with MAC > MACCutoff_to_CollapseUltraRare (=10 by default)
Number_ultra_rare: number of markers that are ultra-rare with MAC <= MACCutoff_to_CollapseUltraRare (=10 by default)
-
A file with association test results for single markers in the set-based tests ** Please Refer to the SAIGE (Single-variant test) Step 2 output files for details. ** For binary traits, effect sizes of single variants can be estimated more accurately through Firth’s Bias-Reduced Logistic Regression by setting * –is_Firth_beta=TRUE and –pCutoffforFirth=0.01 * NOTE: This option is currently only for the single-variant assoc tests and for Burden test (when the only Burden test is conducted (–r.corr=1)). When the SKAT-O test is conducted (–r.corr=0), the Firth’s Bias-Reduced Logistic Regression is not applied.
** If only Burden tests are performed (–r.corr=1), please use –is_single_in_groupTest=TRUE to output the single-variant assoc tests ** If the SKAT-O tests are performed (–r.corr=0), single-variant assoc test results are automatically output.less -S ./output/genotype_100markers_bgen_groupTest_out_cond.txt.singleAssoc.txt -
A file with marker lists for region/set-based tests (–is_output_markerList_in_groupTest=TRUE)
less -S ./output/genotype_100markers_bgen_groupTest_out.txt.markerList.txt